
PHERI—Phage Host ExploRation Pipeline

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
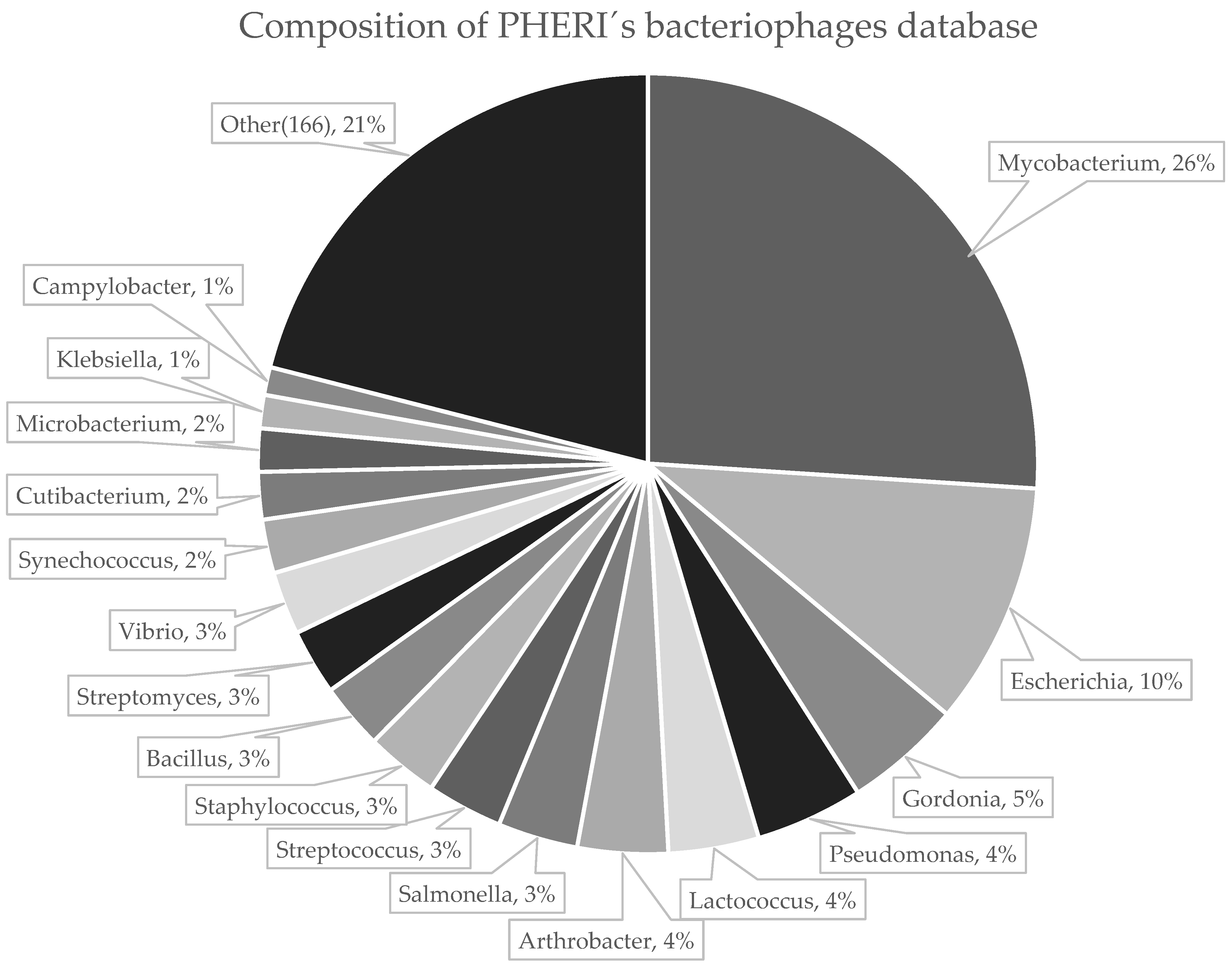
2.1. Collection of Phage Sequences
2.2. Extraction and Annotation of Genes
2.3. Clustering of Gene Sequences
2.4. Training Classification Model
2.5. Classifying Novel Sequence
2.6. Bacterial Strains and Growth Conditions
2.7. Isolation of Bacteriophages
2.8. Plaque Assay and Host Range
2.9. Phage Adsorption
3. Results
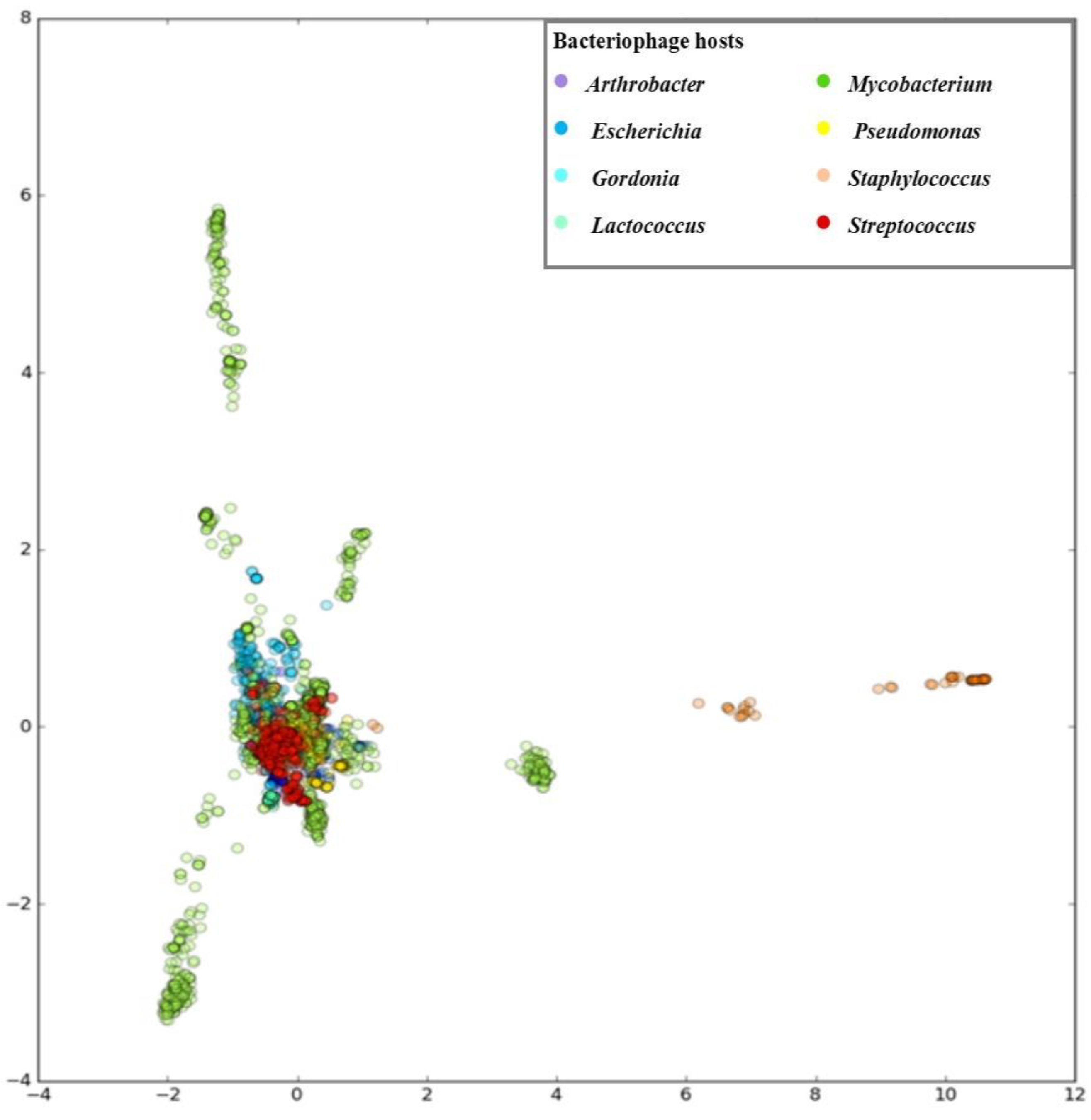
3.1. Developing PHERI Method
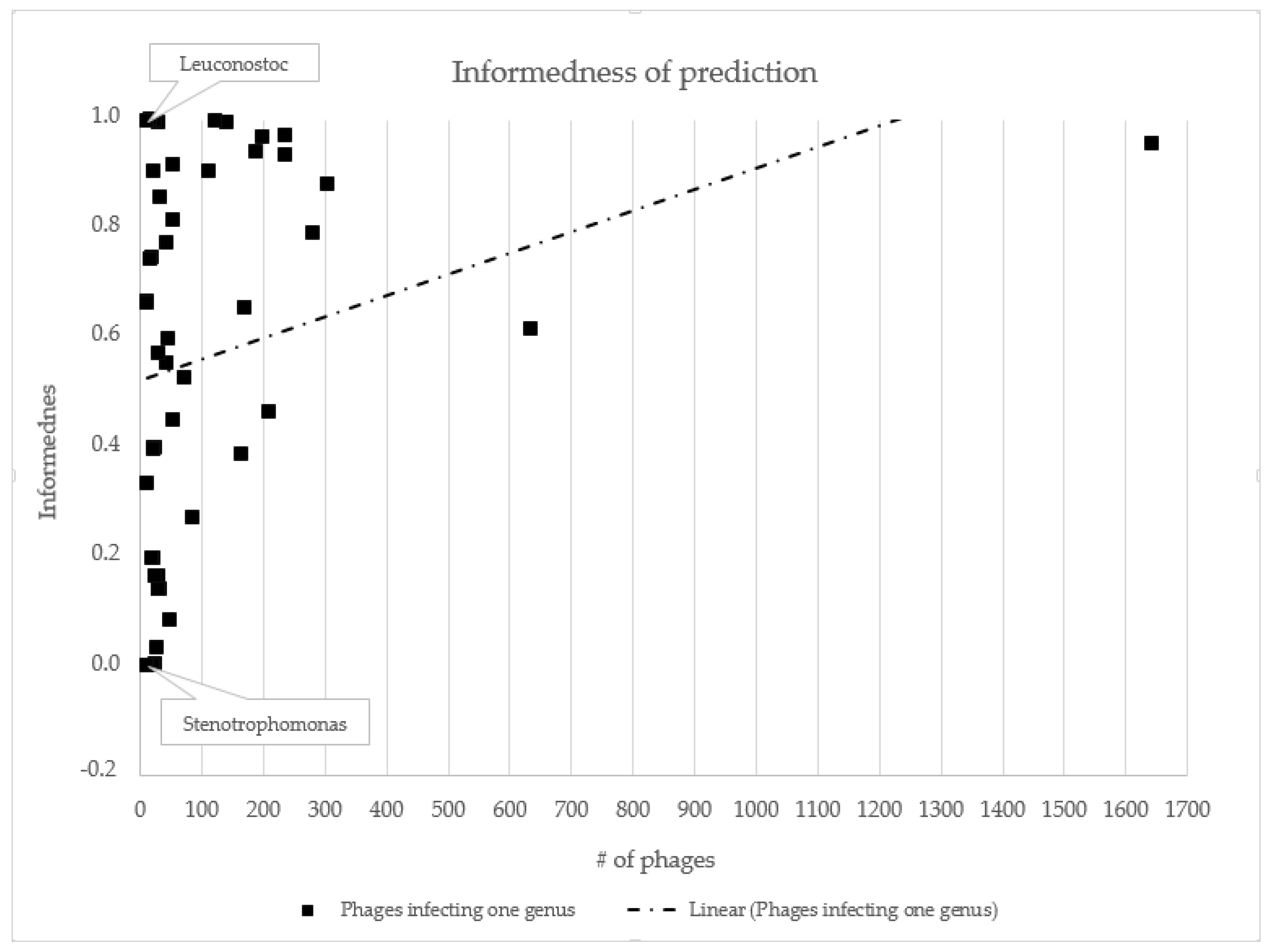
3.2. Host Prediction Evaluation
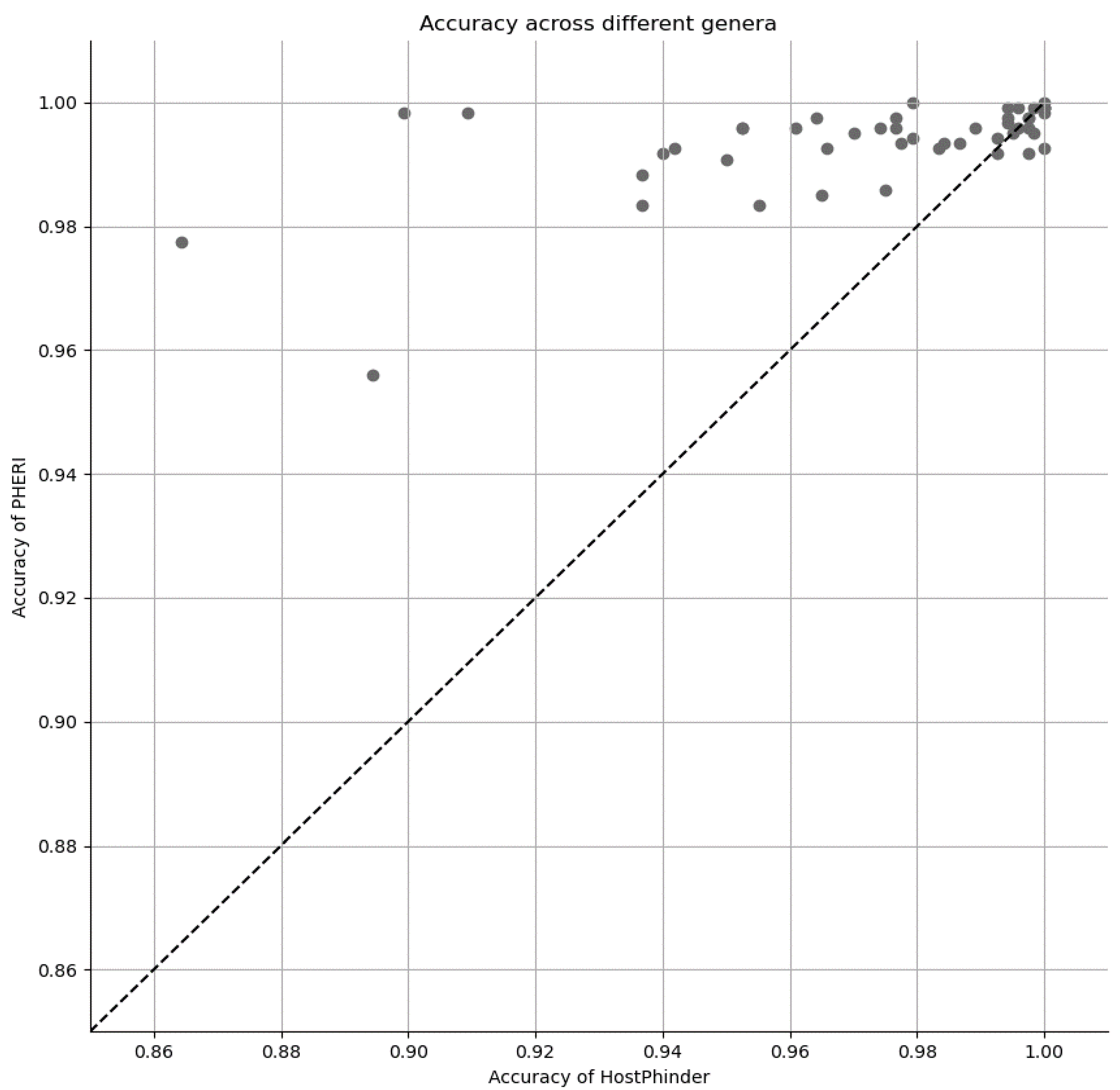
3.3. Comparison of PHERI to Other Tools
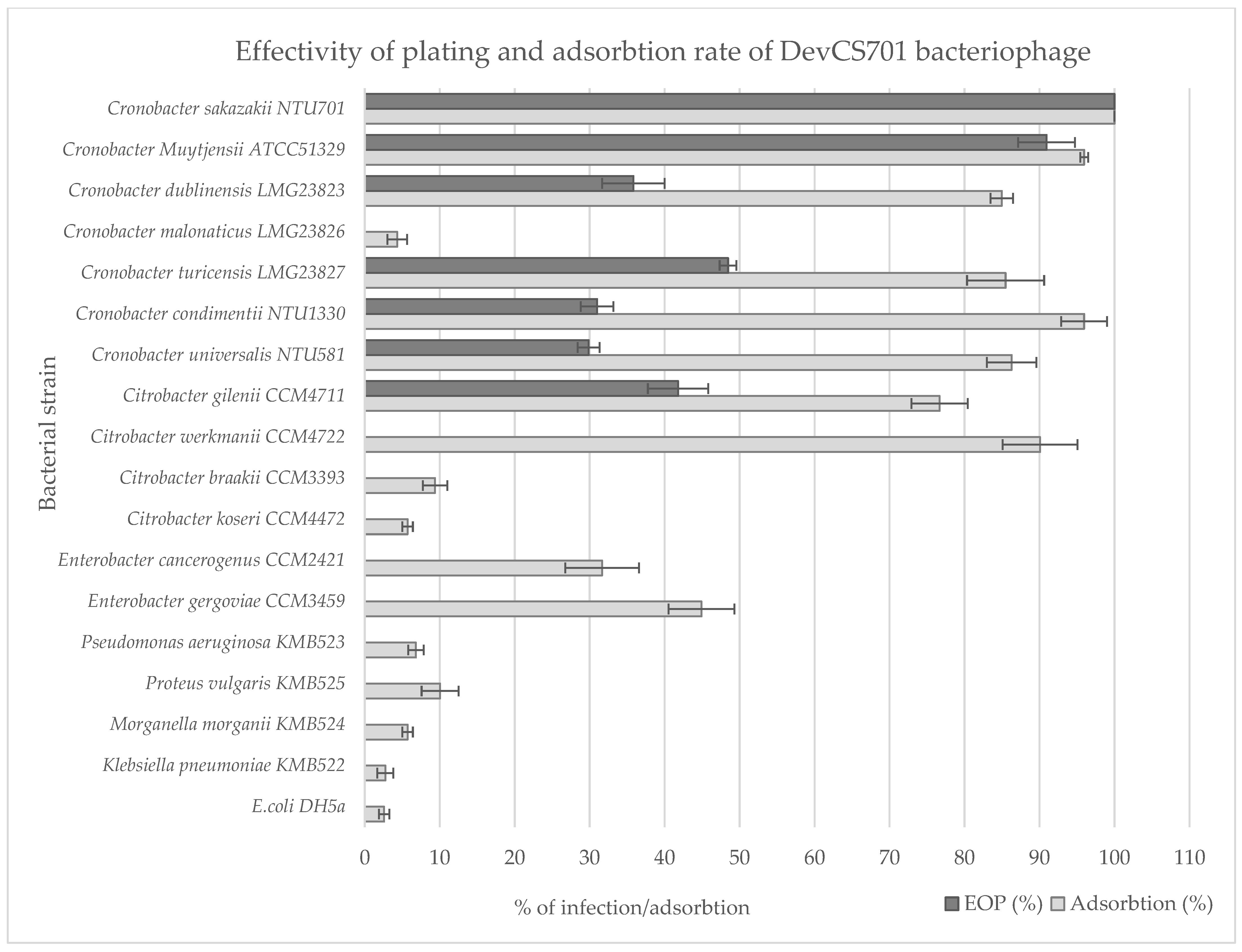
3.4. Host Prediction for New Isolated Bacteriophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ogawara, H. Comparison of Antibiotic Resistance Mechanisms in Antibiotic-Producing and Pathogenic Bacteria. Molecules 2019, 24, 3430. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E. Antibiotics: What Everyone Needs to Know®. In What Everyone Needs to Know(r); Oxford University Press: Oxford, UK, 2019; ISBN 9780190663414. [Google Scholar]
- Williamson, K.E.; Fuhrmann, J.J.; Wommack, K.E.; Radosevich, M. Viruses in Soil Ecosystems: An Unknown Quantity within an Unexplored Territory. Annu. Rev. Virol. 2017, 4, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s Virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; De Roock, S.; Opal, S.M. A Historical Overview of Bacteriophage Therapy as an Alternative to Antibiotics for the Treatment of Bacterial Pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine Viruses–Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Yu, P.; Mathieu, J.; Lu, G.W.; Gabiatti, N.; Alvarez, P.J. Control of Antibiotic-Resistant Bacteria in Activated Sludge Using Polyvalent Phages in Conjunction with a Production Host. Environ. Sci. Technol. Lett. 2017, 4, 137–142. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P. Virus Classification–Where Do You Draw the Line? Arch. Virol. 2018, 163, 2037–2046. [Google Scholar] [CrossRef]
- Yu, P.; Mathieu, J.; Li, M.; Dai, Z.; Alvarez, P.J.J. Isolation of Polyvalent Bacteriophages by Sequential Multiple-Host Approaches. Appl. Environ. Microbiol. 2016, 82, 808–815. [Google Scholar] [CrossRef]
- Ye, M.; Sun, M.; Huang, D.; Zhang, Z.; Zhang, H.; Zhang, S.; Hu, F.; Jiang, X.; Jiao, W. A Review of Bacteriophage Therapy for Pathogenic Bacteria Inactivation in the Soil Environment. Environ. Int. 2019, 129, 488–496. [Google Scholar] [CrossRef]
- Chanishvili, N. Phage Therapy–History from Twort and d’Herelle through Soviet Experience to Current Approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar]
- Myelnikov, D. An Alternative Cure: The Adoption and Survival of Bacteriophage Therapy in the USSR, 1922–1955. J. Hist. Med. Allied Sci. 2018, 73, 385–411. [Google Scholar] [CrossRef]
- Anand, T.; Virmani, N.; Kumar, S.; Kumar Mohanty, A.; Pavulraj, S.; Ch Bera, B.; Vaid, R.K.; Ahlawat, U.; Tripathi, B.N. Phage Therapy for Treatment of Virulent Klebsiella Pneumoniae Infection in Mouse Model. J. Glob. Antimicrob Resist. 2020, 21, 34–41. [Google Scholar] [CrossRef]
- Dissanayake, U.; Ukhanova, M.; Moye, Z.D.; Sulakvelidze, A.; Mai, V. Bacteriophages Reduce Pathogenic Counts in Mice without Distorting Gut Microbiota. Front. Microbiol. 2019, 10, 1984. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B. Effectiveness of Phages in Treating Experimental Escherichia Coli Diarrhoea in Calves, Piglets and Lambs. J. Gen. Microbiol. 1983, 129, 2659–2675. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Huggins, M.B.; Shaw, K.M. Factors Influencing the Survival and Multiplication of Bacteriophages in Calves and in Their Environment. J. Gen. Microbiol. 1987, 133, 1127–1135. [Google Scholar] [CrossRef]
- Carrillo, C.L.; Loc Carrillo, C.; Atterbury, R.J.; El-Shibiny, A.; Connerton, P.L.; Dillon, E.; Scott, A.; Connerton, I.F. Bacteriophage Therapy to Reduce Campylobacter Jejuni Colonization of Broiler Chickens. Appl. Environ. Microbiol. 2005, 71, 6554–6563. [Google Scholar] [CrossRef]
- Cafora, M.; Deflorian, G.; Forti, F.; Ferrari, L.; Binelli, G.; Briani, F.; Ghisotti, D.; Pistocchi, A. Phage Therapy against Pseudomonas Aeruginosa Infections in a Cystic Fibrosis Zebrafish Model. Sci. Rep. 2019, 9, 1527. [Google Scholar] [CrossRef]
- Marza, J.A.S.; Soothill, J.S.; Boydell, P.; Collyns, T.A. Multiplication of Therapeutically Administered Bacteriophages in Pseudomonas Aeruginosa Infected Patients. Burns 2006, 32, 644–646. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and Tolerability of a Cocktail of Bacteriophages to Treat Burn Wounds Infected by Pseudomonas Aeruginosa (PhagoBurn): A Randomised, Controlled, Double-Blind Phase 1/2 Trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Zhvania, P.; Hoyle, N.S.; Nadareishvili, L.; Nizharadze, D.; Kutateladze, M. Phage Therapy in a 16-Year-Old Boy with Netherton Syndrome. Front. Med. 2017, 4, 94. [Google Scholar] [CrossRef]
- Hoyle, N.; Zhvaniya, P.; Balarjishvili, N.; Bolkvadze, D.; Nadareishvili, L.; Nizharadze, D.; Wittmann, J.; Rohde, C.; Kutateladze, M. Phage Therapy against Achromobacter Xylosoxidans Lung Infection in a Patient with Cystic Fibrosis: A Case Report. Res. Microbiol. 2018, 169, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Law, N.; Logan, C.; Yung, G.; Furr, C.-L.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful Adjunctive Use of Bacteriophage Therapy for Treatment of Multidrug-Resistant Pseudomonas Aeruginosa Infection in a Cystic Fibrosis Patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.-L.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M.; et al. Early Clinical Experience of Bacteriophage Therapy in 3 Lung Transplant Recipients. Am. J. Transplant. 2019, 19, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered Bacteriophages for Treatment of a Patient with a Disseminated Drug-Resistant Mycobacterium Abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage Host Range and Bacterial Resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [PubMed]
- Tu, J.; Park, T.; Morado, D.R.; Hughes, K.T.; Molineux, I.J.; Liu, J. Dual Host Specificity of Phage SP6 Is Facilitated by Tailspike Rotation. Virology 2017, 507, 206–215. [Google Scholar] [CrossRef]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine Modifications Protect Phage DNA from Host Restriction Systems. Nat. Commun. 2019, 10, 5442. [Google Scholar] [CrossRef]
- Furi, L.; Crawford, L.A.; Rangel-Pineros, G.; Manso, A.S.; De Ste Croix, M.; Haigh, R.D.; Kwun, M.J.; Engelsen Fjelland, K.; Gilfillan, G.D.; Bentley, S.D.; et al. Methylation Warfare: Interaction of Pneumococcal Bacteriophages with Their Host. J. Bacteriol. 2019, 201, e00370-19. [Google Scholar] [CrossRef]
- Modell, J.W.; Jiang, W.; Marraffini, L.A. CRISPR-Cas Systems Exploit Viral DNA Injection to Establish and Maintain Adaptive Immunity. Nature 2017, 544, 101–104. [Google Scholar] [CrossRef]
- Chopin, M.-C.; Chopin, A.; Bidnenko, E. Phage Abortive Infection in Lactococci: Variations on a Theme. Curr. Opin. Microbiol. 2005, 8, 473–479. [Google Scholar] [CrossRef]
- Chen, B.; Akusobi, C.; Fang, X.; Salmond, G.P.C. Environmental T4-Family Bacteriophages Evolve to Escape Abortive Infection via Multiple Routes in a Bacterial Host Employing “Altruistic Suicide” through Type III Toxin-Antitoxin Systems. Front. Microbiol. 2017, 8, 1006. [Google Scholar] [CrossRef]
- Stanley, S.Y.; Maxwell, K.L. Phage-Encoded Anti-CRISPR Defenses. Annu. Rev. Genet. 2018, 52, 445–464. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Williamson, S.J.; Allen, L.Z.; Lorenzi, H.A.; Fadrosh, D.W.; Brami, D.; Thiagarajan, M.; McCrow, J.P.; Tovchigrechko, A.; Yooseph, S.; Venter, J.C. Metagenomic Exploration of Viruses throughout the Indian Ocean. PLoS ONE 2012, 7, e42047. [Google Scholar] [CrossRef]
- Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A Phage Host Prediction Tool. Viruses 2016, 8, 116. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-Based Prediction of Phage Virion Proteins Using a Support Vector Machine. Front. Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Salisbury, A.; Tsourkas, P.K. A Method for Improving the Accuracy and Efficiency of Bacteriophage Genome Annotation. Int. J. Mol. Sci. 2019, 20, 3391. [Google Scholar] [CrossRef]
- Chibani, C.M.; Meinecke, F.; Farr, A.; Dietrich, S.; Liesegang, H. ClassiPhages 2.0: Sequence-Based Classification of Phages Using Artificial Neural Networks. Available online: https://www.biorxiv.org/content/10.1101/558171v1 (accessed on 2 May 2020).
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated Classification of Tailed Bacteriophages according to Their Neck Organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An Efficient Algorithm for Large-Scale Detection of Protein Families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.; Griffith, O.L. RefSeq (the Reference Sequence Database). In Dictionary of Bioinformatics and Computational Biology; Wiley-Liss: Hoboken, NJ, USA, 2004. [Google Scholar]
- Cosma, C.L.; Sherman, D.R.; Ramakrishnan, L. The Secret Lives of the Pathogenic Mycobacteria. Annu. Rev. Microbiol. 2003, 57, 641–676. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A Hub for Protein Information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Bateman, A. The Pfam Protein Families Database. Nucleic Acids Res. 2000, 28, 263–266. [Google Scholar] [CrossRef]
- Haft, D.H.; Loftus, B.J.; Richardson, D.L.; Yang, F.; Eisen, J.A.; Paulsen, I.T.; White, O. TIGRFAMs: A Protein Family Resource for the Functional Identification of Proteins. Nucleic Acids Res. 2001, 29, 41–43. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Tristão Ramos, R.J.; de Azevedo Martins, A.C.; da Silva Delgado, G.; Ionescu, C.-M.; Ürményi, T.P.; Silva, R.; Koca, J. CrocoBLAST: Running BLAST Efficiently in the Age of next-Generation Sequencing. Bioinformatics 2017, 33, 3648–3651. [Google Scholar] [CrossRef]
- Heringa, J. Needleman-Wunsch Algorithm. In Dictionary of Bioinformatics and Computational Biology; Wiley-Liss: Hoboken, NJ, USA, 2004. [Google Scholar]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef]
- Kajsik, M.; Durovka, P.; Kajsikova, M.; Rusnakova, D.; Szemes, T.; Drahovska, H. Complete Genome Sequence of New Specific Bacteriophage Dev_CS701. Microbiol. Resour. Announc. 2023, 12, e0003423. [Google Scholar] [CrossRef]
- Boulanger, P. Purification of Bacteriophages and SDS-PAGE Analysis of Phage Structural Proteins from Ghost Particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar]
- Garreta, R.; Moncecchi, G. Learning Scikit-Learn: Machine Learning in Python; Packt Publishing Ltd.: Birmingham, UK, 2013; ISBN 9781783281947. [Google Scholar]
- Mahony, J.; McAuliffe, O.; Paul Ross, R.; van Sinderen, D. Bacteriophages as Biocontrol Agents of Food Pathogens. Curr. Opin. Biotechnol. 2011, 22, 157–163. [Google Scholar] [CrossRef]
- Expert round table on acceptance and re-implementation of bacteriophage therapy. Silk Route to the Acceptance and Re-Implementation of Bacteriophage Therapy. Biotechnol. J. 2016, 11, 595–600. [Google Scholar] [CrossRef]
- Kajsík, M.; Bugala, J.; Kadličeková, V.; Szemes, T.; Turňa, J.; Drahovská, H. Characterization of Dev-CD-23823 and Dev-CT57, New Autographivirinae Bacteriophages Infecting Cronobacter Spp. Arch. Virol. 2019, 164, 1383–1391. [Google Scholar] [CrossRef]
- Kajsík, M.; Oslanecová, L.; Szemes, T.; Hýblová, M.; Bilková, A.; Drahovská, H.; Turňa, J. Characterization and Genome Sequence of Dev2, a New T7-like Bacteriophage Infecting Cronobacter Turicensis. Arch. Virol. 2014, 159, 3013–3019. [Google Scholar] [CrossRef]
- Martínez-García, M.; Santos, F.; Moreno-Paz, M.; Parro, V.; Antón, J. Unveiling Viral-Host Interactions within the “Microbial Dark Matter”. Nat. Commun. 2014, 5, 4542. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral Dark Matter and Virus-Host Interactions Resolved from Publicly Available Microbial Genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Kot, W.; Hansen, L.H.; Neve, H.; Hammer, K.; Jacobsen, S.; Pedersen, P.D.; Sørensen, S.J.; Heller, K.J.; Vogensen, F.K. Sequence and Comparative Analysis of Leuconostoc Dairy Bacteriophages. Int. J. Food Microbiol. 2014, 176, 29–37. [Google Scholar] [CrossRef]
- Fan, H.; Huang, Y.; Mi, Z.; Yin, X.; Wang, L.; Fan, H.; Zhang, Z.; An, X.; Chen, J.; Tong, Y. Complete Genome Sequence of IME13, a Stenotrophomonas Maltophilia Bacteriophage with Large Burst Size and Unique Plaque Polymorphism. J. Virol. 2012, 86, 11392–11393. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Q.; Shen, P.; Huang, Y.-P. Isolation and Characterization of a Novel Filamentous Phage from Stenotrophomonas Maltophilia. Arch. Virol. 2012, 157, 1643–1650. [Google Scholar] [CrossRef]
- McCutcheon, J.; Peters, D.; Dennis, J. Identification and Characterization of Type IV Pili as the Cellular Receptor of Broad Host Range Stenotrophomonas Maltophilia Bacteriophages DLP1 and DLP2. Viruses 2018, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.L.; Lynch, K.H.; Stothard, P.; Dennis, J.J. The Isolation and Characterization of Two Stenotrophomonas Maltophilia Bacteriophages Capable of Cross-Taxonomic Order Infectivity. BMC Genom. 2015, 16, 664. [Google Scholar] [CrossRef] [PubMed]
- Endersen, L.; Buttimer, C.; Nevin, E.; Coffey, A.; Neve, H.; Oliveira, H.; Lavigne, R.; O’Mahony, J. Investigating the Biocontrol and Anti-Biofilm Potential of a Three Phage Cocktail against Cronobacter Sakazakii in Different Brands of Infant Formula. Int. J. Food Microbiol. 2017, 253, 1–11. [Google Scholar] [CrossRef]
- McDermott, J.R.; Shao, Q.; O’Leary, C.; Kongari, R.; Liu, M. Complete Genome Sequence of Citrobacter Freundii Myophage Maroon. Microbiol. Resour. Announc. 2019, 8, e01145-19. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | True Positive | True Negative | False Positive | False Negative |
|---|---|---|---|---|
| Leuconostoc | 4 | 1198 | 0 | 0 |
| Ruegeria | 3 | 1197 | 2 | 0 |
| Helicobacter | 6 | 1194 | 2 | 0 |
| Paenibacillus | 6 | 1192 | 4 | 0 |
| Cutibacterium | 25 | 1173 | 4 | 0 |
| Moraxella | 7 | 1190 | 5 | 0 |
| Synechococcus | 29 | 1166 | 7 | 0 |
| Lactococcus | 47 | 1145 | 9 | 1 |
| Streptococcus | 39 | 1152 | 10 | 1 |
| Mycolicibacterium | 320 | 847 | 33 | 2 |
| Staphylococcus | 37 | 1155 | 8 | 2 |
| Arthrobacter | 45 | 1150 | 4 | 3 |
| Rhodococcus | 11 | 1189 | 1 | 1 |
| Microbacterium | 21 | 1171 | 8 | 2 |
| Bacillus | 32 | 1156 | 11 | 3 |
| Gordonia | 55 | 1135 | 5 | 7 |
| Flavobacterium | 6 | 1194 | 1 | 1 |
| Acinetobacter | 9 | 1188 | 3 | 2 |
| Pseudomonas | 46 | 1129 | 16 | 11 |
| Aeromonas | 7 | 1190 | 3 | 2 |
| Corynebacterium | 3 | 1194 | 4 | 1 |
| Caulobacter | 3 | 1193 | 5 | 1 |
| Proteus | 2 | 1199 | 0 | 1 |
| Mannheimia | 2 | 1197 | 2 | 1 |
| Streptomyces | 23 | 1164 | 3 | 12 |
| Escherichia | 82 | 1044 | 31 | 45 |
| Enterococcus | 6 | 1189 | 3 | 4 |
| Listeria | 4 | 1195 | 0 | 3 |
| Erwinia | 5 | 1192 | 1 | 4 |
| Campylobacter | 8 | 1180 | 7 | 7 |
| Salmonella | 20 | 1147 | 13 | 22 |
| Lactobacillus | 5 | 1185 | 6 | 6 |
| Clostridioides | 2 | 1197 | 0 | 3 |
| Clostridium | 2 | 1195 | 2 | 3 |
| Yersinia | 2 | 1192 | 5 | 3 |
| Vibrio | 13 | 1163 | 6 | 20 |
| Rhizobium | 1 | 1198 | 1 | 2 |
| Klebsiella | 5 | 1176 | 8 | 13 |
| Xanthomonas | 1 | 1194 | 3 | 4 |
| Cronobacter | 1 | 1193 | 4 | 4 |
| Pectobacterium | 1 | 1194 | 2 | 5 |
| Pseudoalteromonas | 1 | 1192 | 4 | 5 |
| Brucella | 1 | 1194 | 1 | 6 |
| Ralstonia | 1 | 1193 | 2 | 6 |
| Cellulophaga | 1 | 1193 | 2 | 6 |
| Burkholderia | 1 | 1191 | 4 | 6 |
| Shigella | 1 | 1184 | 7 | 10 |
| Stenotrophomonas | 0 | 1199 | 0 | 3 |
| Citrobacter | 0 | 1196 | 1 | 5 |
| Mycobacterium | 0 | 1196 | 4 | 2 |
| Bacteriophage | Closest Relative | Real Host | PHERI Prediction |
|---|---|---|---|
| Dev-CS701 | vB_CsaM_leB (KX431559.1) | Cronobacter | Citrobacter |
| vB_EcoM_VP1 | vB_EcoM_JS09 (KF582788) | E. coli | Escherichia |
| vB-EcoM_KMB43 | Rb49-like virus (AY343333) | E. coli | Escherichia |
| vB_KpnP_VP3 | KPV811 (KY000081) | Klebsiella | Klebsiella |
| vB_EcoP_VP5 | 64795_ec1 (KU927499) | E. coli | Escherichia |
| PetSE1 | vB_SenS-Ent1 (NC_019539.1) | Salmonella | Salmonella |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baláž, A.; Kajsik, M.; Budiš, J.; Szemes, T.; Turňa, J. PHERI—Phage Host ExploRation Pipeline. Microorganisms 2023, 11, 1398. https://doi.org/10.3390/microorganisms11061398
Baláž A, Kajsik M, Budiš J, Szemes T, Turňa J. PHERI—Phage Host ExploRation Pipeline. Microorganisms. 2023; 11(6):1398. https://doi.org/10.3390/microorganisms11061398
Chicago/Turabian StyleBaláž, Andrej, Michal Kajsik, Jaroslav Budiš, Tomáš Szemes, and Ján Turňa. 2023. "PHERI—Phage Host ExploRation Pipeline" Microorganisms 11, no. 6: 1398. https://doi.org/10.3390/microorganisms11061398
APA StyleBaláž, A., Kajsik, M., Budiš, J., Szemes, T., & Turňa, J. (2023). PHERI—Phage Host ExploRation Pipeline. Microorganisms, 11(6), 1398. https://doi.org/10.3390/microorganisms11061398